fonte: LUSA / Notícias ao minuto
2 Set. 2015
2 Set. 2015
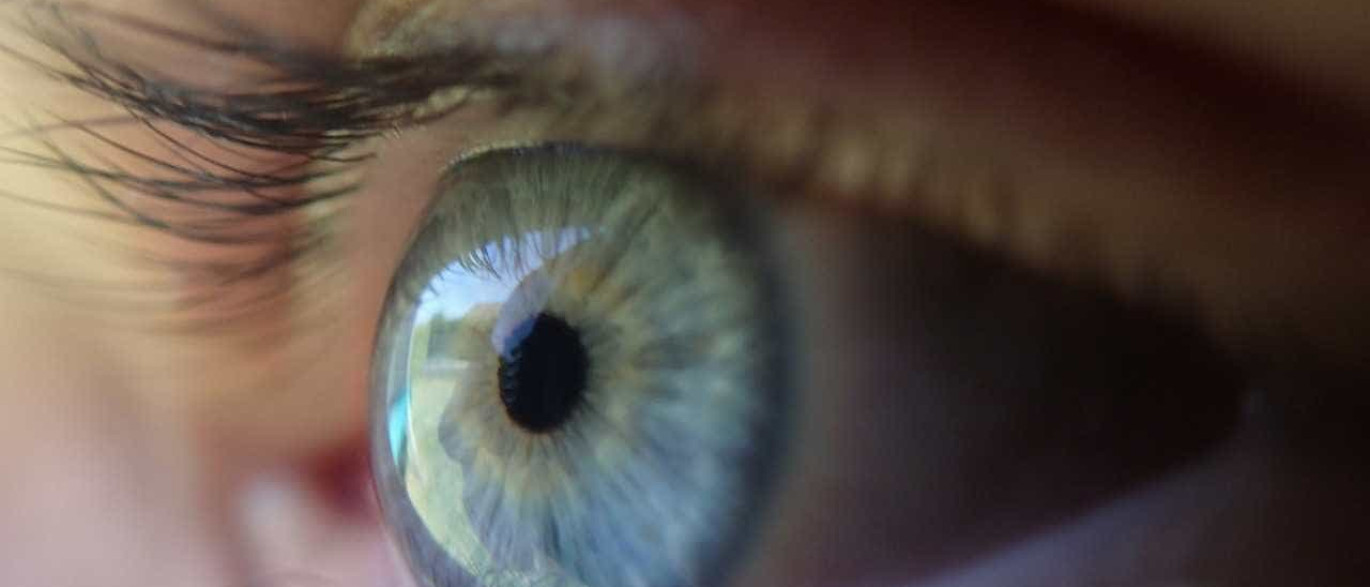
Cientistas descobriram, numa experiência com ratos, que o Meis1 é um dos genes implicados no desenvolvimento correto do olho, durante a fase embrionária, pelo que qualquer disfunção no gene pode gerar microftalmia (globo ocular mais pequeno).
Os resultados da investigação, publicados na revista Development, revelam que o gene Meis1 coordena a expressão de um grande número de genes, por sua vez responsáveis pela formação, pelo desenvolvimento e pela diferenciação da retina neural nos estádios iniciais de formação do olho, assim como de outras partes do organismo.
Os resultados da investigação, publicados na revista Development, revelam que o gene Meis1 coordena a expressão de um grande número de genes, por sua vez responsáveis pela formação, pelo desenvolvimento e pela diferenciação da retina neural nos estádios iniciais de formação do olho, assim como de outras partes do organismo.
"Vimos que a falta do gene Meis1 reduzia muito a formação do esboço do olho. Uma só cópia alterada era suficiente para o conseguir", afirmou, citada hoje pela agência noticiosa Efe, uma das coordenadoras da investigação, Paola Bovolenta, do Centro de Biologia Molecular "Severo Ochoa", em Madrid, Espanha.
Posteriormente, através de análises genómicas, a equipa identificou "os genes alterados na ausência de Meis1" e viu que o gene "coordena um número muito grande de genes implicados no desenvolvimento do olho, que tem um papel fundamental na formação deste órgão e que as suas mutações podem gerar microftalmia", adiantou Paola Bovolenta.
A microftlamia é uma malformação congénita, pouco frequente, que faz com que o olho não se desenvolva bem e que a criança nasça com um ou os dois globos oculares mais pequenos do que o normal e com diversos graus de incapacidade visual, que podem chegar à cegueira.
A anomalia é responsável direta por 11 por cento dos casos de cegueira infantil nos países desenvolvidos, salienta a Efe.
O defeito pode ser hereditário, aparecer de forma espontânea ou ser gerado por agentes externos (como o medicamento Talidomida, comercializado na década de 60 e que provocou uma série de casos de malformações nos fetos). Na maior parte das situações, a microftlamia tem origem em mutações genéticas.
Segundo a investigadora Paola Bovolenta, a microftalmia "também pode vir acompanhada por defeitos na face, no ouvido ou no sistema muscular", uma vez que é provocada "por mutações em genes que são importantes para o desenvolvimento de tecidos distintos".
No estudo participaram igualmente cientistas do Centro Helmholtz de Munique, na Alemanha, e da Universidade da Austrália Ocidental.
Posteriormente, através de análises genómicas, a equipa identificou "os genes alterados na ausência de Meis1" e viu que o gene "coordena um número muito grande de genes implicados no desenvolvimento do olho, que tem um papel fundamental na formação deste órgão e que as suas mutações podem gerar microftalmia", adiantou Paola Bovolenta.
A microftlamia é uma malformação congénita, pouco frequente, que faz com que o olho não se desenvolva bem e que a criança nasça com um ou os dois globos oculares mais pequenos do que o normal e com diversos graus de incapacidade visual, que podem chegar à cegueira.
A anomalia é responsável direta por 11 por cento dos casos de cegueira infantil nos países desenvolvidos, salienta a Efe.
O defeito pode ser hereditário, aparecer de forma espontânea ou ser gerado por agentes externos (como o medicamento Talidomida, comercializado na década de 60 e que provocou uma série de casos de malformações nos fetos). Na maior parte das situações, a microftlamia tem origem em mutações genéticas.
Segundo a investigadora Paola Bovolenta, a microftalmia "também pode vir acompanhada por defeitos na face, no ouvido ou no sistema muscular", uma vez que é provocada "por mutações em genes que são importantes para o desenvolvimento de tecidos distintos".
No estudo participaram igualmente cientistas do Centro Helmholtz de Munique, na Alemanha, e da Universidade da Austrália Ocidental.
Sem comentários:
Enviar um comentário